
Craniosynostosis: The Evolution from Open Craniofacial Techniques to Minimally Invasive Endoscopic Approaches
At the Skull Base Institute, children with craniofacial deformities are seen by a multidisciplinary team of experts including the skull base surgeon, pediatricians, plastic and reconstructive surgeons, geneticists, otolaryngologists, and speech therapists.
These children have congenital deformities of the head, including craniosynostosis, hypertelorism, Apert’s syndrome, Crouzon’s syndrome, and Pfeiffer’s syndrome. The majority are developmentally normal and even those that may be a bit delayed, have the potential to lead normal lives if the deformity is treated in its early stages.
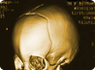
Many of the children have multiple cranial sutures that are permanently fused- preventing the brain from developing properly. These young patients first undergo a sophisticated three-dimensional CT scan. A template – or roadmap – is then made for the complex operation. Surgery involves releasing these sutures and reconstructing the skull into a normal appearance.
The traditional craniofacial procedures to treat craniosynostosis require large exposures and extensive dissection of the soft tissues. At the Skull Base Institute, these surgical procedures have been successfully converted to minimally invasive endoscopic procedures performed through few, small, stab incisions. Sagittal, coronal and metopic craniosynostoses have all been successfully repaired through these endoscopic minimally invasive approaches. These innovative procedures are performed during infancy and are commonly associated with custom designed helmet therapy.
“Congenital” tumors such as teratomas, craniopharyngiomas, neurofibromas and others are similarly resected using advanced minimally invasive endoscopic techniques that are ideally suited for infants and children.
Overview
The term craniosynostosis refers to the premature union (syn-ostosis) of two or more of the bones of the skull (cranium) of an infant’s head. This condition leads to restricted skull growth often resulting in an abnormal head shape.
The infant’s skull is normally made up of free-floating bones which are separated by sutures. This arrangement allows the infant’s head to pass through the birth canal and as an infant’s brain grows, open sutures allow the skull to expand and develop into its normal shape. If one or more of the sutures closes early, it Causes the skull to expand in the direction of the open sutures as the brain is growing normally and will take the path of least resistance which may result in an abnormal head shape. In severe cases, this condition can also cause increased pressure on the growing brain.
The major skull sutures are:
- The coronal sutures, which separate the frontal bone from the parietal bones and run along the top of the skull from side to side in front of the ears
- The metopic suture, or frontal suture, which separates the frontal bones into two halves
- The sagittal suture, which separates the two parietal bones and extends from the anterior fontanelle to the posterior fontanelle or from (bregma) to (lambda)
- The lambdoid sutures, which separate the occipital bones from the parietal bones
Craniosynostosis has been reported to occur approximately once per 1,900 live births, but the true incidence is probably higher. Approximately 63% of all cases occur in males but the male preponderance is 75-80% in sagittal and metopic synostosis (scaphocephaly and trigonocephaly), and the female preponderance is approximately 75% in coronal synostosis (anterior plagiocephaly), and there is a slight female preponderance in (brachycephaly). The frequency of occurrence of the various types of craniosynostosis in the population is approximately as follows: sagittal 50-58%, coronal 20-29%, metopic 4-10%, and lambdoid 2-4%.
Causes
The cause of the premature fusion is unknown. Craniosynostosis may be either:
Primary craniosynostosis; multiple theories have been proposed for the etiology of primary craniosynostosis, but the most widely accepted is a primary defect in ossification of the mesenchymal layer of the cranial bones. Intrauterine factors may also play a role.
Secondary craniosynostosis; more frequent than the primary type and is typically due to an underlying systemic disorder e.g. sickle cell disease, thalassemia, rickets, and all Causes of microcephaly causing primary failure of brain growth and is associated with premature fusion of all of the skull sutures. Secondary craniosynostosis can also develop following placement of a shunt in a child with hydrocephalus (shunt-induced craniosynostosis). The intracranial pressure (ICP) is not elevated in secondary craniosynostosis but the condition is often associated with neurodevelopmental delay.
Simple craniosynostosis is the term given when only one suture fuses prematurely, while complex or compound craniosynostosis is used to describe premature fusion of multiple cranial sutures. Elevated ICP is more common in multiple suture synostosis but rare in single suture synostosis. When children with craniosynostosis, usually complex, also display other body deformities, this is termed syndromic craniosynostosis e.g., Apert’s and Crouzon’s syndromes.
Symptoms
Commonly, craniosynostosis is present at birth, but it may not be always apparent especially in mild cases. Craniosynostosis is evident at birth when associated with other craniofacial abnormalities such as in complex or syndromic craniosynostosis. Primary and secondary craniosynostosis usually become evident as the child grows and his skull takes an abnormal shape or when the child exhibits neurodevelopmental delays.
A variety of skull deformities can be seen in craniosynostosis depending on the number and location of the suture(s) involved, these include:
- Anterior plagiocephaly – Early fusion of one coronal suture
- Posterior plagiocephaly – Early closure of one lambdoid suture
- Scaphocephaly – Early fusion of the sagittal suture
- Trigonocephaly – Early fusion of the metopic suture
- Brachycephaly – Early bilateral coronal suture fusion
Intracranial hypertension, hydrocephalus and mental retardation of varying degrees are all liable to occur in craniosynostosis due to secondary or associated brain malformations, but this is uncommon in single suture (simple) synostosis.
Diagnosis
The initial medical history involves questions about family history of craniosynostosis or other congenital abnormalities, about developmental milestones, as well as questions about abnormal sleeping position and neck tightness (torticollis) as these can cause positional deformities (positional molding), a condition that does not require surgery and is managed conservatively e.g. positional plagiocephaly. Typically, careful physical examination is sufficient to make the diagnosis of craniosynostosis. Physical examination of a craniosynostotic baby involves careful palpation of the skull for suture ridges and soft spots (fontanelles), checking for abnormal neck position (torticollis) and for other deformities in the body. Measurements are also taken of the child’s face and head. The presence of multiple suture fusions strongly suggests a craniofacial syndrome e.g. Apert’s or Crouzon’s. The presence of microcephaly usually suggests a secondary craniosynostosis and its underlying cause should be investigated.
The suspected diagnosis is confirmed by plain X-ray, computerized tomography (CT) scannig and magnetic resonance imaging (MRI). On plain X-ray of the skull the prematurely fused sutures are either not visible or show evidence of sclerosis. Cranial CT scan with bone window is often more informative than plain X-ray especially in diagnosing early suture fusion and in detecting any underlying abnormality of the brain. Cranial CT scan with three-dimensional reconstruction is another useful imaging modality that is of great value in planning the surgical correction when surgery is being considered. MRI is important in showing the anatomy of the brain and the soft tissue structures and in detecting any associated cerebral malformations.
Further studies including the need for genetic counseling, or for ophthalmological, audiological, psychological, or other studies may be recommended according to the type of craniosynostosis and the severity of the condition.
Treatment
Not all children with craniosynostosis require treatment, however, when treatment is indicated, it almost always consists of surgical intervention. The aims of surgery are to allow normal brain growth by providing an adequate space within the skull, to correct increased ICP when it is present, to achieve a cosmetically acceptable head with appropriate shape and dimensions, and to protect the eyes from being affected by the increased ICP or the abnormal configuration of the eye sockets.
The type and timing of surgery varies with the child’s condition and the type of craniosynostosis being treated. The ideal time for surgery, in the absence of increased ICP, is usually between three to six months of age because bones of the skull are easy to work with at that age, and the covering of the brain, known as the dura, can make bone on its own. Early surgery also provides the growing brain with the ideal surroundings and facilitates normal future shaping of the skull. Although surgery is done early in life, there is no upper age limit for surgery. Usually, one surgery is required to correct children with simple craniosynostosis; however, some may need minor alterations at the age of four or five.
Infants with a defined syndrome causing craniosynostosis should be evaluated early for surgery and they may require several surgeries on the skull to achieve a normal shape.
Traditional surgery for craniosynostosis is usually performed via a bicoronal skin flap (across the top of the head from ear to ear). A generous craniectomy to “reopen” the prematurely fused sutures with extension to the adjacent sutures, in addition to restructuring or remodeling of the involved skull bones when required, is the basic procedure.
In positional molding, surgery is seldom indicated and infants improve with repositioning maneuvers and physical therapy for torticollis when present. In severe cases custom-molded helmets are used to correct the skull deformity.
Prognosis
Most patients respond very well to surgical treatment of craniosynostosis and the improvement in head shape is appreciated almost immediately after the operation. The key to treating craniosynostosis is early detection and treatment. If untreated, craniosynostosis results in head deformity that can be severe and permanent. Raised ICP, seizures, vision problems, and developmental delay can occur. In addition to correcting functional problems associated with craniosynostosis, reconstructive surgery has a positive effect on the child’s self image and the ability to get along with his or her peers.
All patients with craniosynostosis must be monitored after surgery with regular measurement of the head circumference and monitoring of the child’s functional development to ensure normal brain growth.
If you or someone you love has questions about Craniosynostosis, please Contact Us and we will be happy to help you.